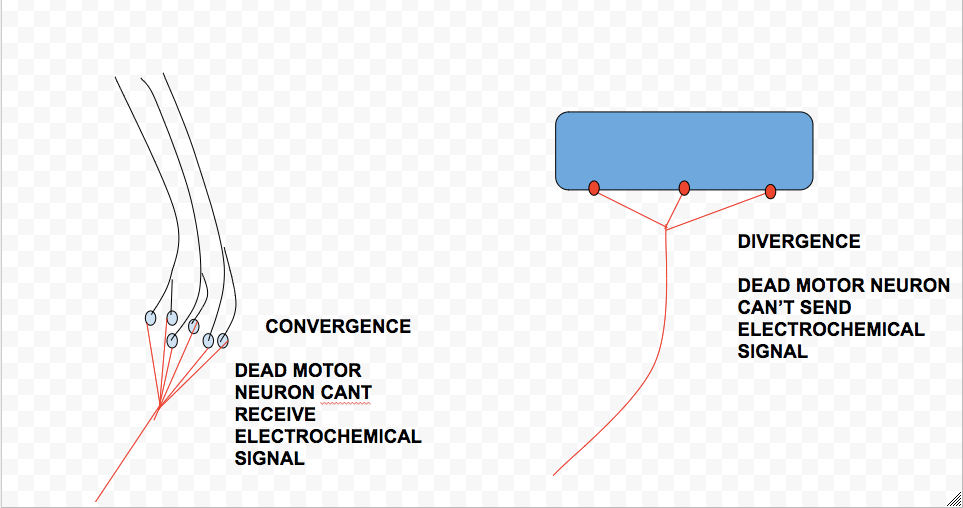
In typical cases of ALS, motor neurons cells in both the motor cortex of the brain and motor neurons in the spinal cord die, although the disease may, sometimes, extend itself to other regions of the brain. Although the cause of ALS is still, relatively speaking, a mystery, one of the defining features of a patient afflicted by the degenerative disease is the TDP-43 protein in the form of an inclusion body. Inclusion bodies are found to “clog up” the cytoplasm of a neuron. ALS can also be inherited, allowing neurologists to examine the genes responsible for the symptoms in order to determine a cure (and also a sense of the disease’s origin). The genes found to be associated with the disease are observed to be related to “…protein degradation, RNA processing, and the [cell’s] cytoskeleton…” (Wikipedia). Furthermore, Excitotoxicity, a high concentration of calcium in neurons, has been observed to play a role in the death of them; likewise, neurofilament accumulation has been observed in a few cases. Ultimately, with motor neurons dead, the body is not able to convert a stimulus propagated by sensory neurons into an action as the receiver of this signal, the motor neurons, become unresponsive. Furthermore, these neurons are no longer able to carry out a muscular response as their death inhibits them from distributing any electrochemical signal elsewhere.
For this post, I used https://en.wikipedia.org/wiki/Amyotrophic_lateral_sclerosis, specifically the pathophysiology section as well as Chapter 2 in our textbook Nerve Cells, Neural Circuitry, and Behavior by Eric R. Kandel, Ben A. Barres, and A.J. Hudspeth.
